Research
The research in the Guo Laboratory is centered on the structural biology of amyloid-related neurodegenerative diseases. In particular, we are interested in the structure and mechanism of protein aggregation involved in Alzheimer’s disease and prion disease.
Alzheimer’s Disease
Alzheimer’s disease, the most common type of dementia, is a progressive and fatal neurodegenerative disorder, with no definitive cause and no cure. In the world, the number of people living with dementia is estimated to be 47 million in 2015, and will reach 135 million in 2050. The major risk factor is age. It rarely occurs in people under the age of 65. In contrast, almost half of the people over age 85 have dementia.
The prevailing paradigm for Alzheimer’s disease pathogenesis is the amyloid hypothesis, which contends that accumulation of amyloid-β (Aβ) protein in the forms of non-fibrillar or fibrillar aggregates initiates a cascade of events leading to neuronal death and ultimately the clinical symptoms of Alzheimer’s disease. The fibrillar aggregates are called “amyloid”. The Guo lab has been working to understand the process of Aβ aggregation and the structure of various fibrillar and non-fibrillar Aβ aggregates.
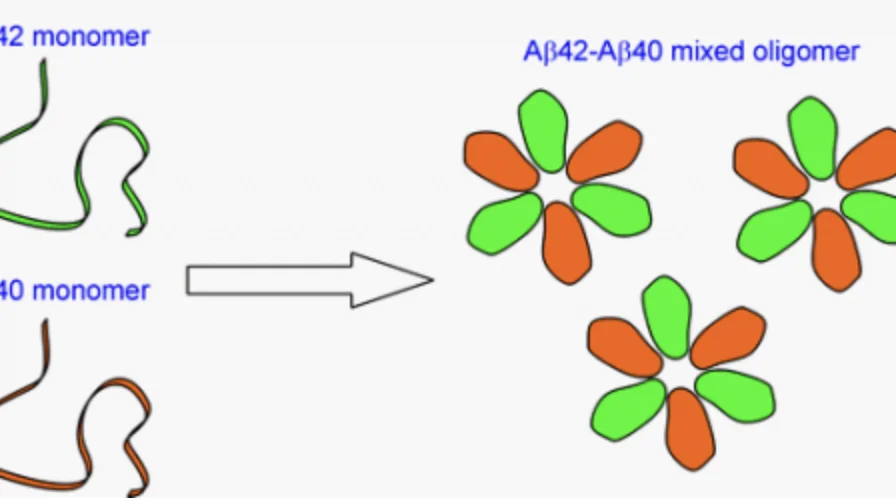
Alzheimer’s Aβ42 and Aβ40 form mixed oligomers with direct molecular interactions
Our Areas Of Focus
Structure of Aβ Oligomers
Aβ42 oligomers are believed to be the toxic species in Alzheimer’s disease. Inhibitors of oligomer toxicity may prove to be effective treatments of Alzheimer’s disease.
One approach would be high throughput screening of available compound libraries, but this approach is limited by what is already available. The Guo lab is taking a structural biology approach by studying the structure of Aβ oligomers. With detailed structural information, we will be able to rationally design inhibitors that target specific structures. Aβ oligomers are known to be heterogeneous, with many different oligomers identified mostly based on immunoreaction to conformation-specific antibodies.
With the structural approach, the Guo lab aims to characterize different Aβ oligomers and then to design inhibitors that target different oligomer structures. The strength of the Guo lab is our electron paramagnetic resonance (EPR) technique. EPR does not require crystals (necessary for X-ray crystallography) and can tolerate heterogeneity (which causes problems for NMR) because only one distance is studied at a time. Using EPR, we have shown that Aβ42 adopt anti-parallel β structures in oligomers, in contrast to parallel β structures of protein fibrils.
A major effort of current drug development is to target amyloid plaques, which consist of fibrils with parallel structures, and these experimental drugs will not inhibit oligomers due to their structural difference. This fact may partly explain the recent failures of clinical trials for several experimental drugs. Our goal is to obtain high resolution structural models of Aβ42 oligomers to facilitate drug development targeting oligomers.
Structure of Aβ Fibrils
Aβ fibril is the major component of amyloid plaques in Alzheimer’s disease. Although plaques are generally considered to be non-toxic, they can catalyze the formation of oligomers through secondary nucleation and they also serve as a reservoir of Aβ monomers. Aβ fibril is also a major drug target in clinical trials. Structural knowledge of Aβ fibrils is critical for understanding the relationship between oligomers and fibrils, and also for structure-based drug design targeting Aβ fibrils.
The Guo lab has studied the structure of Aβ40 fibrils under two experimental conditions. Aβ40 forms two different fibril morphologies under these two conditions. EPR data reveal structural differences of these two fibril forms, and provide a mechanistic understanding of fibril polymorphism. Currently, we are investigating the structure of Aβ42 fibrils using spin labeling and EPR.
Mechanism of Aβ Aggregation
Aβ protein is a small fragment of a much larger protein called amyloid precursor protein (APP), which is cleaved sequentially by two enzymes, β-secretase and g-secretase, to produce Aβ. The mechanism by which Aβ aggregates to form oligomers and fibrils is critical for identifying different points of therapeutic intervention, and how different intervention may affect the dynamics of various Aβ species.
One major effort in this area is focused on the interactions between Aβ40 and Aβ42, two major Aβ species in the brain. The only difference between Aβ40 and Aβ42 is that Aβ42 has two extra amino acids at the carboxyl terminus. In the brain, Aβ40 concentration is approximately 10 times more than that of Aβ42. However, the plaques, one of the pathological hallmarks of Alzheimer’s disease, consist of mostly Aβ42. Aβ40 is present in only about one third of the plaques, suggesting that Aβ42 deposition in the brain precedes Aβ40 aggregation. Soluble Aβ42 levels in Alzheimer’s patients are found to be lower than normally aged individuals, while Aβ40 levels show no difference between Alzheimer’s and normal populations. This is consistent with the preferential deposition of Aβ42 in the brain. Using EPR, we have shown that Aβ40 and Aβ42 form interlaced fibrils. We are currently investigating the interactions between Aβ40 and Aβ42 during the process of aggregation.
Another effort of the Guo lab is to identify different Aβ species during the early aggregation process and study its structure with EPR. We are using aggregation kinetics to reveal different steps of aggregation. Strategies to isolate different Aβ species is being developed in order to study their structures with EPR.
Prion Disease
Prion disease is caused by an infectious agent that is composed of only protein. Prion protein exists in two conformations, a disease conformation called PrPSc ('Sc' for scrapie) that causes disease, and a normal conformation called PrPC. PrPSc is able to induce conformational changes in PrPC and converts PrPC to PrPSc. Proteins that show prion-like behavior have also been found in fungi, including HET-s in filamentous fungus Podospora anserina, Sup35, Ure2, and others in yeast Saccharomyces cerevisiae. Our lab is interested in studying the fungal prion proteins as a model system to elucidate the mechanism of prion disease.
Structure of Yeast Prion Ure2 Fibrils
Yeast prions, despite differences in sequence from their mammalian counterparts, share similar features with mammalian prions including infectivity, prion strain phenomenon, and species barrier. Therefore, yeast prions are good model systems for human prion diseases. Structural knowledge on yeast prion fibrils is important for understanding the structural basis of prion propagation and the prion strain phenomenon.
The Guo lab is interested in the structural characterization of yeast prion protein Ure2 fibrils using EPR. We have shown that Ure2 amyloid fibrils adopt a hierarchical organization in their amyloid core. The inner core show stronger packing interactions than the outer core region as judged by the strength of spin-spin interactions. Furthermore, we demonstrated a strategy to identify secondary structure using quantitative analysis of spin exchange interactions. Currently, we are working to obtain high resolution structural models for the Ure2 fibrils formed under different experimental conditions.
Our Projects
Mutagenesis Screening
Investigating the effect that mutations have on the structure of aggregation proteins: Tau, IAPP, and alpha-synuclein.
Aβ Aggregation
Using electron paramagnetic resonance technique to characterize the structure of Aβ oligomers.
Fyn
Incorporating the fyn protein - a molecule involved in several biochemical pathways in the brain - into the lactamase binding assay.
Lactamase Binding
Designing a split beta-lactamase reporter assay with high specificity to relate protein aggregation to lactamase activity.
Database
Analyzing available data and track AB protein levels in patients with neurodegenerative diseases, such as Alzheimer's, over time.
Screening
Running high throughput screening on libraries of 200,000+ compounds to test their effects on aggregation.